처음 R로 RNA-seq 분석했을 때 삽질 기록
RNA-seq 분석을 R로 처음 시도하면서 겪었던 시행착오와 삽질의 기록. FASTQ부터 DESeq2까지, 아무도 안 알려주는 실전 팁을 정리했습니다.
시작은 단순했다: "RNA-seq 분석해볼까?"
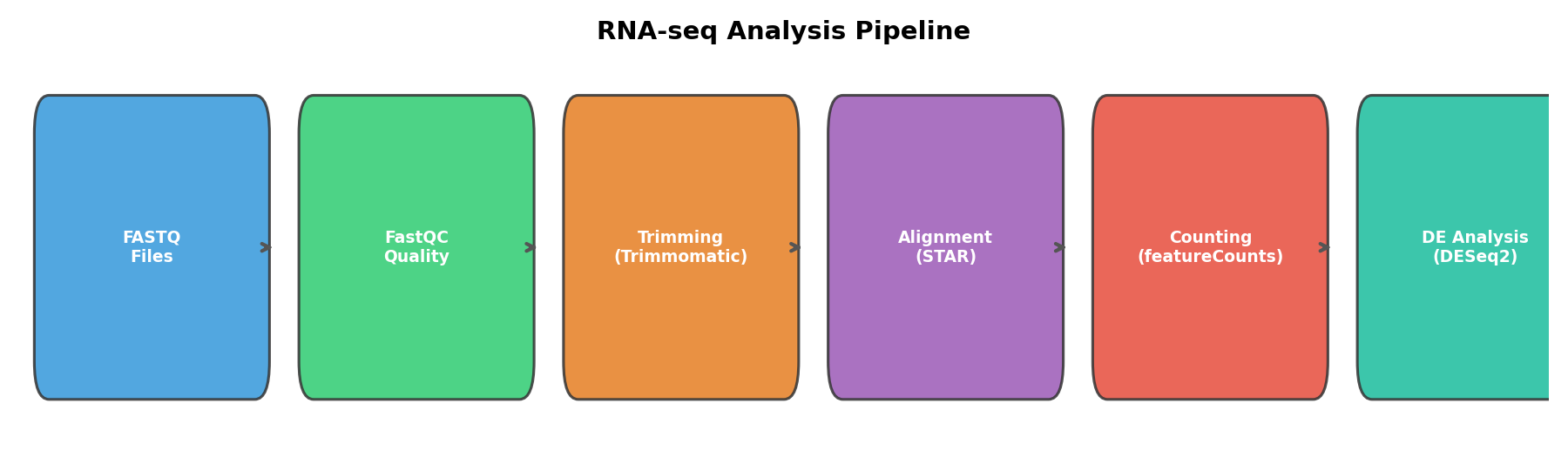
솔직히 말하면, 처음 RNA-seq 분석을 시작했을 때 내가 뭘 모르는지도 몰랐다. 교수님이 "이 FASTQ 파일로 differential expression 분석 좀 해봐"라고 하셨을 때, 나는 구글에 "RNA-seq analysis R tutorial"을 검색하는 것부터 시작했다. 지금 생각하면 그 때의 나에게 해주고 싶은 말이 한 트럭이다.
논문에서는 Methods 섹션에 "DESeq2를 사용하여 분석했다"고 딱 한 줄인데, 그 한 줄 뒤에 숨겨진 삽질의 양은 상상을 초월한다. 이 글은 그 삽질의 솔직한 기록이다.
FASTQ 파일을 처음 봤을 때의 충격
터미널을 열고 head 명령어로 FASTQ 파일을 열어봤는데, 의미 불명의 문자열이 쏟아져 나왔다. Quality score가 ASCII로 인코딩되어 있다는 걸 알기까지 꽤 걸렸다. Phred33이니 Phred64니 하는 개념도 처음에는 전혀 감이 안 왔다.
@SRR123456.1 1 length=150
ATCGATCGATCGATCG...
+
FFFFFFFF:FFFFFF...
처음에는 이걸 R에서 직접 읽으려고 했다. readLines()로 FASTQ를 열어서 파싱하려 했던 건 지금 생각해도 부끄럽다. 당연히 메모리가 터졌다. 40GB짜리 파일을 R에서 텍스트로 읽겠다니.
alignment 도구 선택에서의 혼란
STAR를 쓸지 HISAT2를 쓸지 고민하다가 일주일을 날렸다. 각종 벤치마크 논문을 읽었는데, 결론은 "둘 다 괜찮다"였다. 결국 STAR를 골랐는데, 이유는 단순했다 — 연구실 선배가 STAR 스크립트를 갖고 있었기 때문이다.
문제는 genome index를 만드는 과정이었다. 처음에 --sjdbOverhang 파라미터를 read length - 1로 안 맞추고 기본값으로 돌렸더니, 나중에 결과가 이상해서 다시 다 돌려야 했다. 레퍼런스 genome 다운로드부터 index 생성까지, 그냥 이 과정만 사흘이 걸렸다.
# 이걸 몰라서 한참 헤맸다
STAR --runMode genomeGenerate \
--genomeDir /path/to/star_index \
--genomeFastaFiles GRCh38.primary_assembly.genome.fa \
--sjdbGTFfile gencode.v38.annotation.gtf \
--sjdbOverhang 149 \
--runThreadN 16
NCBI의 GEO 데이터베이스에서 공개 데이터를 받아서 연습했는데, SRA toolkit의 fastq-dump이 또 한 세월이었다. 나중에야 fasterq-dump이 있다는 걸 알았다.
R로 넘어오면서 진짜 시작된 삽질
alignment까지는 어찌어찌 했는데, R에서 count matrix를 만드는 과정에서 또 막혔다. featureCounts로 만든 결과를 R로 읽어들이는 것까지는 좋았다. 문제는 그 다음이었다.
DESeq2 설치부터 난관
# 이 한 줄이 왜 이렇게 어려운지
BiocManager::install("DESeq2")
처음에는 install.packages("DESeq2")로 설치하려 했다. 당연히 안 된다. Bioconductor에서 설치해야 한다는 걸 알고 BiocManager를 깔았는데, 의존성 패키지 설치에서 에러가 줄줄이 나왔다. XML 라이브러리가 없다, openssl 버전이 안 맞다... 시스템 라이브러리를 apt-get으로 깔아야 한다는 걸 몰랐다.
삽질 끝에 알게 된 건, R 패키지 설치 에러의 90%는 시스템 레벨 라이브러리 문제라는 것이다.
count matrix의 함정
DESeqDataSetFromMatrix에 넣을 count matrix를 만들 때, 컬럼 순서가 sample information과 안 맞아서 결과가 완전히 뒤집어진 적이 있다. Control과 Treatment의 라벨이 바뀌어서, fold change가 정반대로 나왔는데, 처음에는 "오 이 유전자가 이렇게 올라가는구나" 하고 넘어갔었다.
# 이 확인을 안 해서 큰일 날 뻔했다
all(colnames(count_matrix) == rownames(sample_info))
# FALSE... 😱
이걸 발견한 건 순전히 운이었다. 알려진 marker gene의 발현 방향이 문헌과 반대라서 뭔가 이상하다 싶어 처음부터 다시 확인했더니 발견됐다. 데이터 sanity check의 중요성을 뼈저리게 느낀 순간이었다.
normalization, 도대체 뭘 써야 하나
RPKM, FPKM, TPM, CPM... 처음에는 이 약자들이 다 같은 건 줄 알았다. Excel에서 각 유전자의 count를 total read count로 나누면 되는 거 아닌가? 라고 생각했던 과거의 나를 때리고 싶다.
DESeq2는 자체 normalization (median of ratios)을 쓴다는 걸 알게 된 후에도, "그러면 TPM은 언제 쓰는 거지?"라는 의문이 한동안 해결되지 않았다. 결론은:
- differential expression: DESeq2 내부 normalization 사용 (raw count 넣기)
- 시각화/비교: TPM이나 VST-transformed 값 사용
- 절대로 하면 안 되는 것: normalized 값을 DESeq2에 넣기
이 구분을 명확히 이해하기까지 Bioconductor support forum의 질문글을 수십 개는 읽었다. Mike Love (DESeq2 개발자)의 답변이 정말 많은 도움이 됐다.
p-value vs adjusted p-value, 그리고 volcano plot
결과가 나왔을 때 처음에는 p-value < 0.05인 유전자가 수천 개 나와서 "와 대박, 이렇게 많이 차이나나?" 했다. Multiple testing correction을 모르고 raw p-value만 본 거다.
adjusted p-value (FDR)를 적용하니까 유의미한 유전자가 확 줄었다. 처음에는 실망했는데, 이게 정상이라는 걸 나중에 알았다.
# 결과 확인 — padj를 봐야 한다!
res <- results(dds, alpha = 0.05)
summary(res)
# volcano plot — 이거 처음 그렸을 때의 감동
library(EnhancedVolcano)
EnhancedVolcano(res,
lab = rownames(res),
x = 'log2FoldChange',
y = 'pvalue',
title = 'Treatment vs Control')
처음으로 volcano plot을 그렸을 때의 그 감동은 아직도 기억난다. 내가 만든 데이터에서 나온 그림이 논문에서 보던 것과 비슷하게 생겨서 진짜 신기했다.
재현성 — 가장 뼈아픈 교훈
한 달 후에 같은 분석을 다시 돌렸는데 결과가 달랐다. 패키지 버전이 업데이트되면서 기본 파라미터가 바뀐 거였다. sessionInfo()를 저장해두라는 조언을 무시했던 게 화근이었다.
그 후로는 모든 분석에 renv를 사용하고, 분석 시작할 때 반드시 환경을 기록한다.
# 제발 이것만은 해두자
writeLines(capture.output(sessionInfo()), "sessionInfo.txt")
# 더 좋은 방법
renv::init()
renv::snapshot()
요즘은 바이오인포매틱스 분석의 재현성이 업계에서도 큰 화두인데, BioAI Market(sysofti.com)에서 관련 도구들의 트렌드를 확인해보면 Docker 기반 파이프라인이 빠르게 늘고 있다.
돌이켜보며 — 삽질이 실력이 된다
지금은 RNA-seq 분석을 꽤 편하게 할 수 있지만, 처음의 그 삽질이 없었으면 지금의 이해도가 없었을 것이다. 튜토리얼을 따라하기만 하면 "돌아가긴 하는데 왜 이렇게 하는지 모르는" 상태가 된다. 삽질하면서 에러를 만나고, 원인을 찾고, 해결하는 과정에서 진짜 실력이 쌓인다.
유전체 데이터 분석이 건강과 의료에 미치는 영향이 점점 커지고 있다. GenoBalance(genobalance.com)에서 유전체 기반 건강 정보를 다루고 있는데, 이런 분석의 임상 적용 사례를 보면 우리가 하는 기초 분석이 얼마나 중요한지 새삼 느끼게 된다.
입문자에게 전하는 팁 요약
- FASTQ → BAM → Count 파이프라인은 쉘 스크립트로 먼저 익히자
- DESeq2에는 raw count만 넣자. 절대 normalized 값 넣지 말 것
- sample info와 count matrix의 순서를 반드시 확인하자
sessionInfo()저장은 필수,renv는 강추- p-value가 아니라 adjusted p-value를 봐야 한다
- Stack Overflow와 Bioconductor support를 친구로 삼자
- 삽질은 부끄러운 게 아니다. 삽질 기록을 안 남기는 게 부끄러운 거다
이 글이 RNA-seq 분석을 처음 시작하는 누군가에게 "아 나만 이런 게 아니구나"라는 위안이 되었으면 좋겠다. 다음에는 Bioconductor 패키지 선택에 대해서도 경험을 공유할 예정이다.
관련 리소스:
관련 글
Can an LLM Run an RNA-seq Analysis on Its Own? Building ARIA, a Decision-Aware Transcriptome Framework
6월 2일 · 6 min read
BioinformaticsDESeq2 vs edgeR vs limma-voom: Complete Comparison for RNA-seq Differential Expression
5월 4일 · 12 min read
BioinformaticsBioconductor 패키지 고르는 현실적인 기준
2월 18일 · 11 min read
ProteomicsHandling Missing Values in Proteomics: Imputation Methods Compared
2월 25일 · 6 min read