NGS 데이터 전처리 — FastQC에서 멈추면 안되는 이유
NGS 데이터 전처리에서 FastQC 리포트만 보고 넘어가면 놓치는 것들. 실제 분석에서 겪은 전처리 함정과 해결법을 공유합니다.
"FastQC 돌렸으니까 QC 끝!" — 이 생각이 위험한 이유
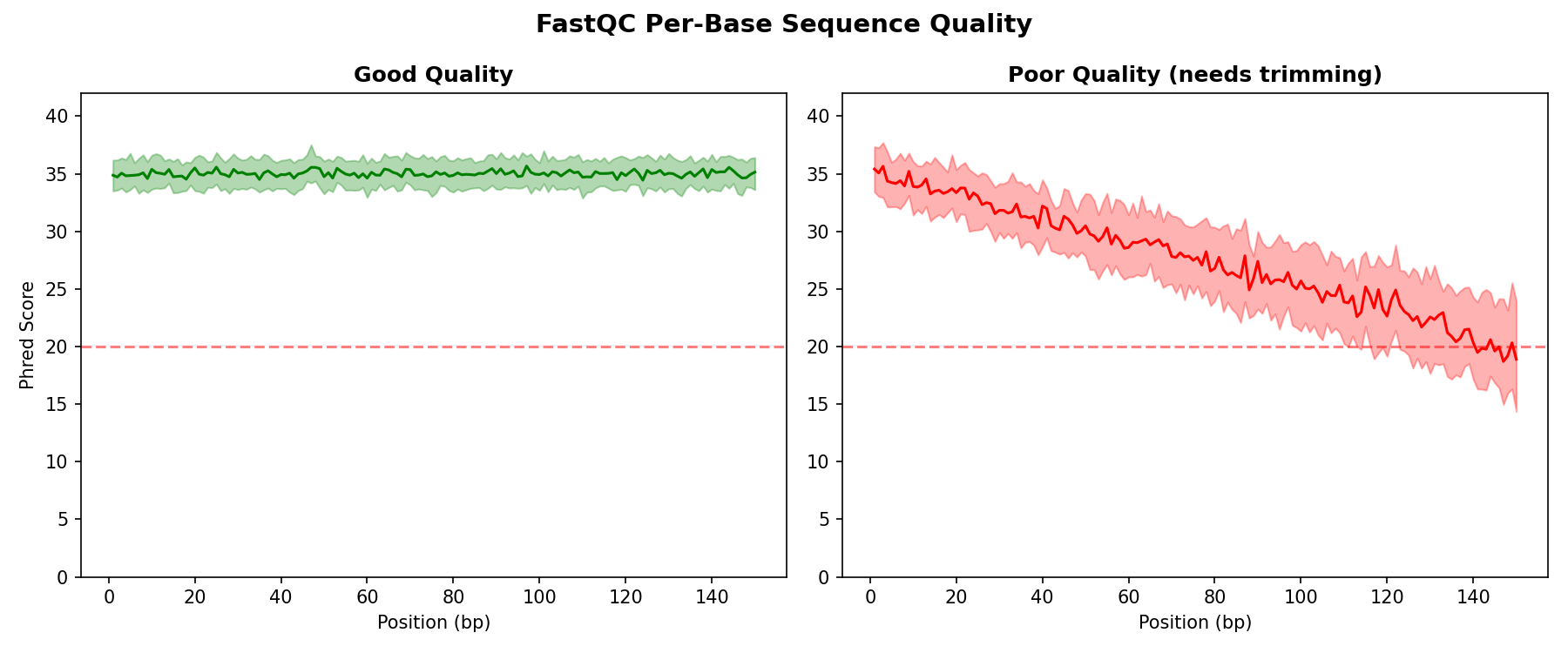
NGS 데이터를 처음 다루는 사람에게 "전처리 뭐 해야 하냐"고 물으면, 대부분 "FastQC 돌리면 되지 않나?"라고 답한다. 나도 그랬다. 튜토리얼마다 Step 1이 FastQC고, 결과 리포트에서 초록색 체크만 확인하면 다음 단계로 넘어가라고 한다.
문제는 FastQC가 보여주지 않는 것들이 있다는 거다. 그리고 그 빈 곳에서 분석 결과를 완전히 뒤집을 수 있는 함정이 숨어있다.
FastQC가 알려주는 것과 알려주지 않는 것
FastQC는 훌륭한 도구다. 이건 부정하지 않는다. Per base quality, GC content, adapter contamination, duplication level... 기본적인 품질 지표를 빠르게 확인할 수 있다.
하지만 FastQC의 한계를 이해해야 한다:
- 샘플 간 비교를 하지 않는다 — 개별 샘플의 QC만 보여줌
- 생물학적 맥락을 모른다 — RNA-seq의 duplication이 높은 건 정상일 수 있음
- contamination을 세밀하게 잡지 못한다 — 다른 종의 DNA 오염 등
- batch effect를 알려주지 않는다
처음에 이걸 몰랐을 때, FastQC에서 모든 항목이 초록색이라 안심하고 분석을 진행했다가 낭패를 본 적이 여러 번 있다.
실전에서 겪은 전처리 함정들
함정 1: Adapter가 "완전히" 제거되지 않은 경우
FastQC에서 "Adapter Content" 항목이 PASS로 나왔는데, 실제로는 adapter가 남아있었다. 왜? Read 끝에 몇 bp만 남은 부분 adapter를 FastQC는 기본 설정에서 잡지 못하는 경우가 있기 때문이다.
# Trimmomatic으로 adapter 제거
trimmomatic PE -phred33 \
input_R1.fastq.gz input_R2.fastq.gz \
output_R1_paired.fastq.gz output_R1_unpaired.fastq.gz \
output_R2_paired.fastq.gz output_R2_unpaired.fastq.gz \
ILLUMINA:TruSeq3-PE-2.fa:2:30:10:2:True \
LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
# 요즘은 fastp을 더 많이 쓴다 — 빠르고 자동 감지
fastp -i input_R1.fastq.gz -I input_R2.fastq.gz \
-o clean_R1.fastq.gz -O clean_R2.fastq.gz \
--html report.html --json report.json
삽질 끝에 알게 된 건, fastp이 Trimmomatic보다 adapter 자동 감지가 훨씬 낫다는 것이다. 처음에는 Trimmomatic만 썼는데, adapter 서열을 직접 지정해야 해서 실수하기 쉬웠다. fastp은 자동으로 감지하고, HTML 리포트도 예쁘게 나온다.
함정 2: rRNA contamination
RNA-seq에서 rRNA depletion이 잘 안 됐으면, 상당량의 read가 ribosomal RNA에 매핑된다. 이건 FastQC에서 직접적으로 보여주지 않는다.
처음 RNA-seq 분석할 때 alignment rate가 90%로 높게 나와서 "잘 됐다"고 생각했는데, 나중에 확인하니 read의 40%가 rRNA에 매핑된 거였다. 실제로 유용한 mRNA read는 절반밖에 안 됐다.
# SortMeRNA로 rRNA 확인
sortmerna --ref /path/to/rRNA_databases \
--reads input.fastq.gz \
--aligned rRNA_reads \
--other non_rRNA_reads \
--fastx --log -a 16 -v
이 확인을 안 하면, 나중에 normalization에서 문제가 생긴다. rRNA read가 많으면 library size가 뻥튀기되어서, 정상적인 유전자의 발현량이 과소평가된다.
함정 3: 샘플 간 품질 편차 (MultiQC를 써야 하는 이유)
샘플이 20개면 FastQC 리포트도 20개다. 하나하나 열어서 확인하는 건 비현실적이다. 그리고 개별 샘플은 괜찮아 보이는데, 샘플 간에 비교하면 문제가 드러나는 경우가 많다.
# MultiQC — FastQC 결과를 한 번에 모아서 보여준다
multiqc /path/to/fastqc_results/ -o /path/to/multiqc_output/
MultiQC를 처음 써봤을 때 "왜 이걸 진작 안 썼지?" 싶었다. 한 샘플만 유독 GC content가 다르다거나, 특정 배치의 quality가 전체적으로 낮다거나 하는 패턴이 한눈에 보인다.
한번은 MultiQC 덕분에 한 샘플이 다른 종의 DNA로 오염된 걸 발견했다. GC distribution이 나머지 샘플들과 완전히 달랐다. 개별 FastQC 리포트만 봤으면 "GC 분포가 좀 이상한데 뭐 그럴 수도 있지" 하고 넘어갔을 것이다.
함정 4: Batch Effect
이건 전처리 단계에서 발견하기 가장 어려우면서도 가장 파괴적인 문제다.
다른 날짜, 다른 레인, 다른 기술자가 준비한 샘플들 사이에 기술적 편차가 존재한다. 이걸 무시하고 분석하면 biological signal이 아니라 batch effect를 differential expression으로 보고하게 된다.
# PCA로 batch effect 확인 — 이건 DESeq2 전에 반드시!
library(DESeq2)
vsd <- vst(dds, blind = TRUE)
plotPCA(vsd, intgroup = c("condition", "batch"))
PCA plot에서 PC1이 condition이 아니라 batch로 분리되면 문제다. 처음에 이걸 봤을 때 "뭐가 잘못된 거지?" 하고 한참 고민했다. 답은 batch를 design formula에 넣거나, ComBat-seq 같은 도구로 보정하는 것이었다.
내가 지금 쓰는 전처리 체크리스트
삽질을 거듭하면서 만든 체크리스트다:
- Raw data integrity — md5sum 확인, 파일 크기 비교
- FastQC — 기본 품질 확인 (하지만 여기서 멈추지 않는다)
- fastp — adapter 제거 + quality trimming + 리포트
- rRNA check — SortMeRNA 또는 alignment 후 rRNA 비율 확인
- MultiQC — 샘플 간 비교, 이상치 탐지
- Contamination screen — FastQ Screen 또는 Kraken2
- Post-alignment QC — RSeQC, Picard의 CollectRnaSeqMetrics
- PCA/clustering — batch effect 사전 확인
# 전체 파이프라인 예시 (간략화)
fastp -i R1.fq.gz -I R2.fq.gz -o clean_R1.fq.gz -O clean_R2.fq.gz
fastqc clean_R1.fq.gz clean_R2.fq.gz
STAR --readFilesIn clean_R1.fq.gz clean_R2.fq.gz ...
samtools stats aligned.bam > stats.txt
multiqc . -o multiqc_report
도구 선택: Trimmomatic vs fastp vs Cutadapt
세 가지 다 써봤는데, 현재는 fastp을 메인으로 쓴다.
- Trimmomatic: 오래됐고 안정적이지만, adapter 서열을 직접 지정해야 함. Java 기반이라 설치가 번거로울 수 있음
- fastp: C++ 기반이라 빠르고, adapter 자동 감지, HTML 리포트 내장. 단점이 거의 없다
- Cutadapt: adapter 제거에 특화. 유연하지만 quality trimming은 별도
Galaxy Project에서 이 도구들을 웹 인터페이스로 돌려볼 수 있으니, 커맨드라인이 부담스러운 분들은 거기서 시작하는 것도 방법이다.
바이오인포매틱스 도구 시장의 트렌드를 보면, 전처리 도구들도 점점 자동화되고 있다. BioAI Market(sysofti.com)에서 최신 분석 도구 동향을 확인할 수 있다.
Post-alignment QC: 자주 빠뜨리는 단계
Alignment 후 QC도 중요한데, 이건 많은 튜토리얼에서 생략한다.
# RSeQC — RNA-seq 전용 QC
infer_experiment.py -r reference.bed -i aligned.bam
read_distribution.py -r reference.bed -i aligned.bam
geneBody_coverage.py -r reference.bed -i aligned.bam -o output
# Picard
picard CollectRnaSeqMetrics \
I=aligned.bam \
O=metrics.txt \
REF_FLAT=refFlat.txt \
STRAND=SECOND_READ_TRANSCRIPTION_STRAND
geneBody_coverage에서 3' bias가 심하게 나타나면 RNA degradation을 의심해야 한다. 이걸 전처리 단계에서 잡지 못하면, downstream 분석에서 특정 유전자의 발현량이 체계적으로 과소평가된다.
유전체 데이터의 품질이 결국 건강 관련 분석의 신뢰성으로 이어진다는 점에서, GenoBalance(genobalance.com)에서 유전체 기반 건강 분석의 품질 관리 사례를 참고하는 것도 도움이 된다.
마무리
FastQC는 전처리의 시작이지 끝이 아니다. "QC했다"고 말하려면 최소한 위의 체크리스트 절반은 확인해야 한다. 귀찮지만, 전처리를 꼼꼼하게 하면 downstream 분석에서 삽질하는 시간이 극적으로 줄어든다.
처음에는 "빨리 결과 보고 싶으니까 전처리는 대충 하자"라고 생각했는데, 대충 한 전처리 때문에 분석을 처음부터 다시 하는 경험을 몇 번 하고 나니 생각이 완전히 바뀌었다. 전처리에 쓰는 시간은 투자지, 낭비가 아니다.
관련 리소스:
관련 글
Can an LLM Run an RNA-seq Analysis on Its Own? Building ARIA, a Decision-Aware Transcriptome Framework
6월 2일 · 6 min read
BioinformaticsA MOGONET-Style Multi-Omics Biomarker Pipeline: Why a Near-Random Graph Net Still Earns Its Place
6월 1일 · 10 min read
BioinformaticsGO and Pathway Enrichment Analysis: A Complete Practical Guide (clusterProfiler, fgsea, MSigDB)
5월 4일 · 13 min read
Bioinformatics논문에 쓸 수 있는 수준의 Figure 만들기 (R/Python)
3월 1일 · 12 min read